NMR
We first became involved with solid-state 13C NMR in the late 1980s as a means of examining structure in complex polymer systems that could not be solubilised without destruction. This approach is certainly useful for lignin but in the case of polysaccharides, we more often do NMR experiments to probe their conformation and molecular rigidity. Almost all our solid-state NMR experiments are run on the EPSRC spectrometer at Durham, where we have a long-standing collaboration with David Apperley.
Solid-state 13C NMR spectra of cell walls, obtained by the standard CP-MAS (cross-polarisation, magic-angle spinning) method, show in principle one peak for each carbon atom in each polymer. In practice, because the peaks are much broader than in solution-state NMR many of the peaks overlap but more or less well-separated diagnostic signals are obtained from the two carbons involved in the glycosidic bond (e.g. C-1 and C-4 for cellulose or pectic galacturonan) and for the exocyclic carbon if any (C-6 of glucose or galactose, C-5 of arabinofuranose). The position in the spectrum (chemical shift) of these carbons is conformation-dependent and can be used to deduce chain conformations, at least on an empirical basis. The underlying theory at present is good for a-linked polysaccharides, doubtful for b linkages and non-existent for C-6, but that does not stop the empirical correlations from being useful. As an example of this approach, the 21 helical (eggbox) and 31 helical forms of pectic galacturonan give distinctive C-4 signals at 77 and 80 ppm respectively: both can be found in cell-wall spectra and the ratio appears to vary with hydration.
We have used solid-state NMR extensively to
estimate how rigid any polymer is within the composite structure of the cell
wall. There are a number of types of experiment in which this can be done. To
summarise:
Proton relaxation times, measured for individual polymers through the 13C spectrum, give an estimate of molecular mobility that is averaged over spatial distances of the order of nanometers by proton spin diffusion. (in fact this phenomenon can sometimes be used to estimate the distance between two polymers on the nm scale) Of the assorted relaxation times that can be measured, we find the most useful are the 1H rotating-frame spin-lattice relaxation time T1r, especially for rigid polymers like cellulose, and the 1H spin-spin relaxation time T2 which is more useful for rather mobile polysaccharides.
The kinetics of cross-polarisation in hydrated cell
walls can be harnessed to discriminate very mobile polysaccharides such as
pectic galactans. These experiments are rather tricky and do not succeed on all
spectrometers: some of our collaborators have not been able to get them
working, but they run well on the Varian system at Durham.
13C
relaxation parameters, particularly the 13C spin-lattice relaxation
time T1, can be measured in direct-polarisation (DP-MAS)
experiments and give specific data on individual carbons: there is almost no
spatial averaging. However these experiments are not so easy to do. In a
conventional 13C T1 experiment on cell walls, the
rigid cellulose carbons relax so slowly that it needs an extremely long
experiment to build up enough signal/noise. Alternatively, slowly-relaxing
signals can be eliminated by saturation using a short recycle time, but if
conventional high-power proton decoupling is used this experiment is very
unkind to the proton channel transmitter, and expensive damage is likely. The
solution is to use multiple-pulse proton decoupling of one kind or another, and
we have found that by judicious choice of the decoupling pulse sequence we can
tune this approach to polymers in specific mobility classes. The spectrum below
was obtained with WALTZ-16 proton decoupling and picks out the arabinan
side-chains of pectin, even though the galactan side-chains are of nearly as
high mobility and are present, in that sample, at greater abundance. The
arabinan signals show strong motional line-narrowing as conformational
broadening is averaged out, indicating mean chain conformations essentially
identical with those found in aqueous solution. We have coined the term
'tethered liquid' for this type of nanophase.
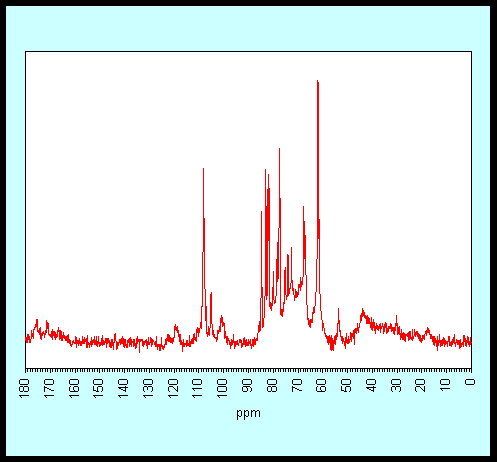
DP-MAS
spectrum of Citrus cell walls, with WALTZ-16 proton decoupling.
Signal assignments as follows:
|
Polymer |
Carbon |
ppm |
|
Galacturonan |
C-6 (free) |
175.4 |
|
Galacturonan |
C-6 (ester) |
171.4 |
|
Arabinan |
C-1 (t-) |
109.3 |
|
Arabinan |
C-1 (5-) |
108.2 |
|
Arabinan |
C-1 (3,5-) |
107.8 |
|
Galactan |
C-1 |
105.1 |
|
Galacturonan |
C-1 |
100.8 |
|
Arabinan |
C-4 (t-) |
84.7 |
|
Arabinan |
C-4 (5-/3,5-) |
83.0 |
|
Arabinan |
C-2 (5-/3,5-) |
82.1 |
|
Arabinan |
C-2 (t-) |
81.6 |
|
Arabinan |
C-3 (3,5-) |
80.0 |
|
Galactan |
C-4 |
78.4 |
|
Arabinan |
C-3 (t-, 5-) |
77.4 |
|
Galactan |
C-2/C-3/C-5 |
75.2 |
|
Galactan |
C-2/C-3/C-5 |
74.0 |
|
Galactan |
C-2/C-3/C-5 |
72.6 |
|
Arabinan |
C-5 (5-) |
67.7 |
|
Arabinan |
C-5 (3,5-) |
67.2 |
|
Arabinan |
C-5 (t-) |
62.0 |
|
Galactan |
C-6 |
61.5 |
|
Galacturonan |
OCH3 |
53.6 |
|
Rhamnose |
CH3 |
17.6 |