Absorption corrections in WinGX
1. Introduction
WinGX contains a number of options for absorption
corrections [1], most of which are examined in the comparison
below. The refined parameters principally affected by the systematic
errors due by absorption are the anisotropic thermal parameters,
more appropriately called anisotropic displacement parameters (adp's)
[see ref 2 and also this site].
Since absorption primarily affects the low angle data,
the net result is that the Uij tensor refines to a smaller
value than the "true" value. In severe cases, it may
even result in negative eigenvalues (i.e. a non-positive
definite Uij tensor).
There are three basic methodologies for applying absorption corrections
to reflection data. These are laid out in the menu scheme in WinGX,
in decreasing order of theoretical rigour.
- Exact numerical corrections - analytical, Gaussian quadrature,
spherical and cylindrical
- Semi-empirical corrections - psi-scans, CAMEL-JOCKEY and multiscan
- Refined corrections - DIFABS, XABS2, SHELXA
2. Numerical methods
It is generally agreed that the best absorption corrections are
provided by the analytical [3] or Gaussian quadrature [4] methods.
These two methods require that the crystal faces are well defined
and can be accurately indexed and measured. These conditions are
not often met. It can be time consuming to index a specimen with
numerous faces and difficult to measure accurately the distances
between faces. For these reasons, numerical methods are used less
often than those which are easier to implement experimentally.
In cases of severe absorption however, they are the only really effective
methods.
The spherical and cyclindrical corrections also provide numerically
accurate corrections for crystals adopting the requisite morphology,
but are less useful, since grown crystals are not
usually spheres or cylinders. Crystals may be ground to spheres, but many materials
do not survive such treatment, The program
XtalView is provided in WinGX as an aid to
face-indexing.
3. Semi-empirical methods
The semi-empirical methods rely on further intensity measurements.
The multiscan method of Blessing [5] is of most use when there
is a large redundancy in the data-set, as is usually the case
for area-detector data. Equivalent intensities are analysed in
terms of a multipolar spherical harmonic expansion and the method
is implemented in the programs PLATON [6], SORTAV [7] and SADABS
[8]. A somewhat similar method called CAMEL-JOCKEY [9] uses a
trigonometric series expansion of the diffractometer angles, but
is little used since so many more experimental measurements are
required. The most commonly used method is the azimuthal scan
or psi-scan method of North et al [10]. This is the simplest
method, involving the measurement of the intensities of a (usually
small) number of reflections with chi values close to 90o
at different psi(phi) values. An averaged absorption surface is thus computed and used
to calculate the transmission factors. It works remarkably well,
but is unsuitable for crystals with large muR values.
4. Refined corrections
The final type of absorption corrections, the so-called refined
corrections DIFABS [11], XABS2 [12] and SHELXA [13] have fallen
out of favour in the recent past. For comments on the use of DIFABS see
the web site
http://www.unige.ch/crystal/stxnews/stx/discuss/dis-dif2.htm
These methods rely on a refined model
being available (hence the structure must be solved) and calculate
the absorption surface from the differences between the observed
and calculated structure factors. The various programs differ
in the exact mathematical functions used to model these differences,
but all suffer from the same philosophical problem, in that the
data is being modified to fit the model. One way round this philosophical
problem would be to incorporate the parameters into a least-squares
refinement.
5. A comparison of methods using NAWO4
A data-set for sodium tungstate dihydrate
(Na2WO4.2H2O)
is provided (download NAWO4 here) for WinGX as test data for absorption corrections.
The compound was chosen because :
- it is commercially available in crystalline form
- the crystal used for the data collection has reasonable but
not perfect facets so it may serve as an example of a "real"
crystal specimen
- the linear absorption coefficient is very large
- only one heavily absorbing atom is present in the asymmetric
unit
Crystal data for Na2WO4.2H2O : a = 8.4797(5) b =
10.5930(5) c = 13.8527 (10) Å, V = 1244.33(1)
Å3, orthorhombic, space group Pbca, Z = 8, Mr
= 329.8, T = 295 K, mu(Mo-K-alpha) = 18.664 mm-1, crystal size
0.391x0.344x0.109 mm. 7543 reflections were measured, yielding 1811
unique reflections.
All refinements were carried out using SHELXL-93 and a number
of criteria for assessing the results are given. The transmission
factors obtained are given in Table 1 and the results of refinement
are summarised in Tables 2 and 3. As expected, all methods offer
a significant improvement over the uncorrected data in terms of
the residuals. The best methods are clearly the analytical and
Gaussian quadrature, which give virtually identical results. For
all other methods, (except XABS2) the range of transmission factors
is smaller, suggesting they may be under-correcting the data.
The method which gives the second best set of figures in Table
2 is DIFABS. At this stage it is also useful to compare the adp's
especially the degree of anisotropy given by the three principal
mean-square displacements. The eigenvalues of the Uij
tensors
of the W and Na atoms are listed in Table 3. The accepted wisdom
is that absorption errors cause the adp's to be somewhat smaller
and more anisotropic than the "true" values (in this
context the "true" value is assumed to be that obtained
from the analytical correction.). Examination of the values obtained
with no absorption correction bear this out. It can be seen that
the adp for the W atom calculated using DIFABS is slightly smaller
and more isotropic than the "true" value, but the agreement
is quite respectable. Moreover the adp's for the light Na atoms
are also quite similar to their "true" value. These
results suggest that the
oft quoted problems with DIFABS are not
always manifest. Rather unexpectedly in view of the large muR
value, the psi-scan method gives values in Table 2 quite similar
to DIFABS. However, the resultant adp's are much less acceptable.
None of the remaining methods provide a satisfactory correction
to the NAWO4 data. The next best correction is probably SHELXA
(though it does give a large +ve residual), followed by multiscan
and XABS2. The multiscan method of Blessing [5] surprisingly performs
rather poorly for the NAWO4 data, even though there is a large
degree of redundancy. It may be that the transmission paths have
not been sampled adequately in this example.
The analytical and DIFABS corrections are further compared in Table 4. The
W-O bond lengths can be seen to be insensitive to the method of correction,
in particular the values using DIFABS and the analytical correction differ
by 2xsigma at most.
The ORTEP views of the WO4 anion are shown below (click on thumb-nail
to view full size picture).
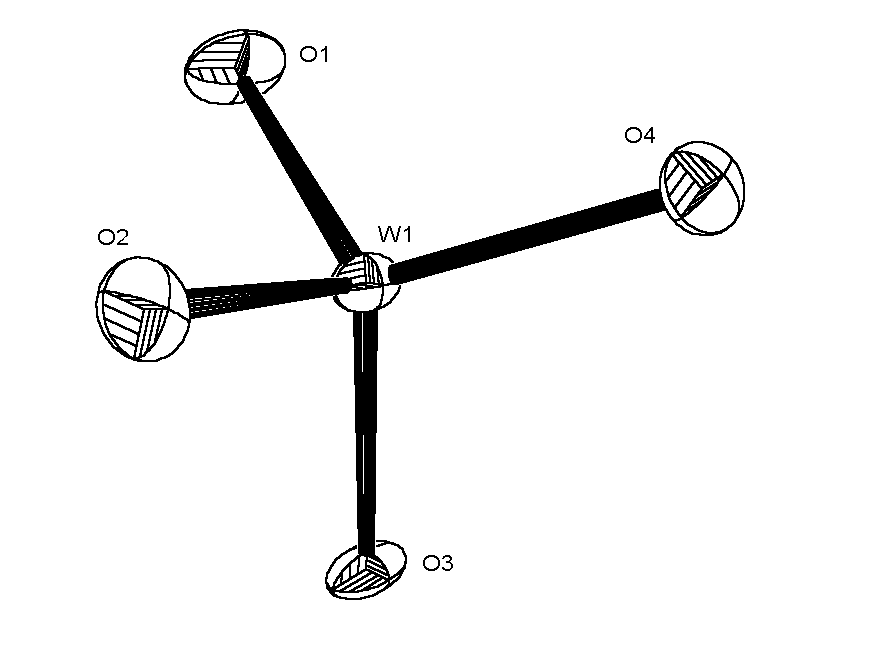
|
ORTEP view of WO4 anion with NO absorption correction.
|
|
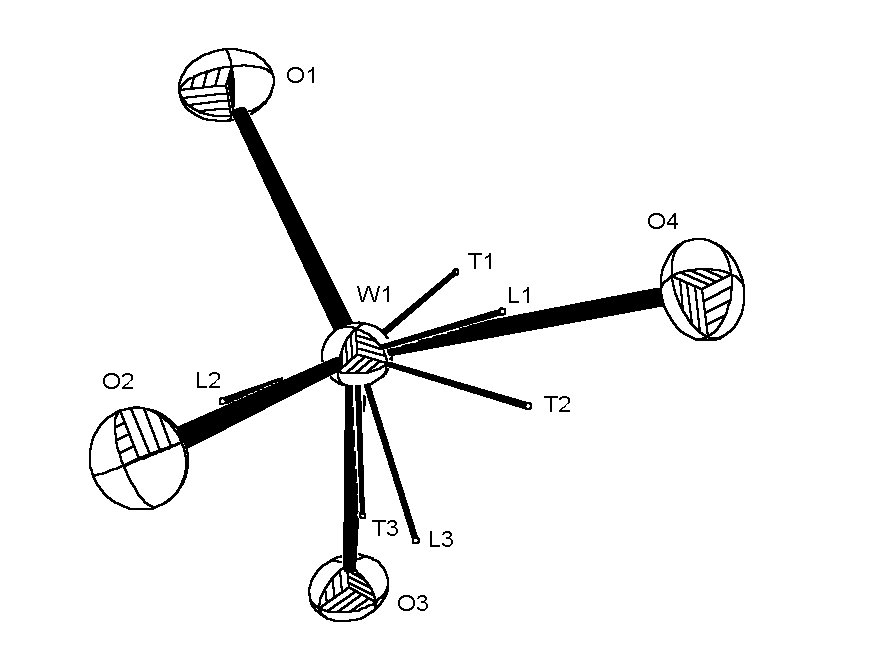
|
ORTEP view of WO4 anion with analytical absorption correction,
showing the principal axes of the libration and translation tensors T and L.
|
|
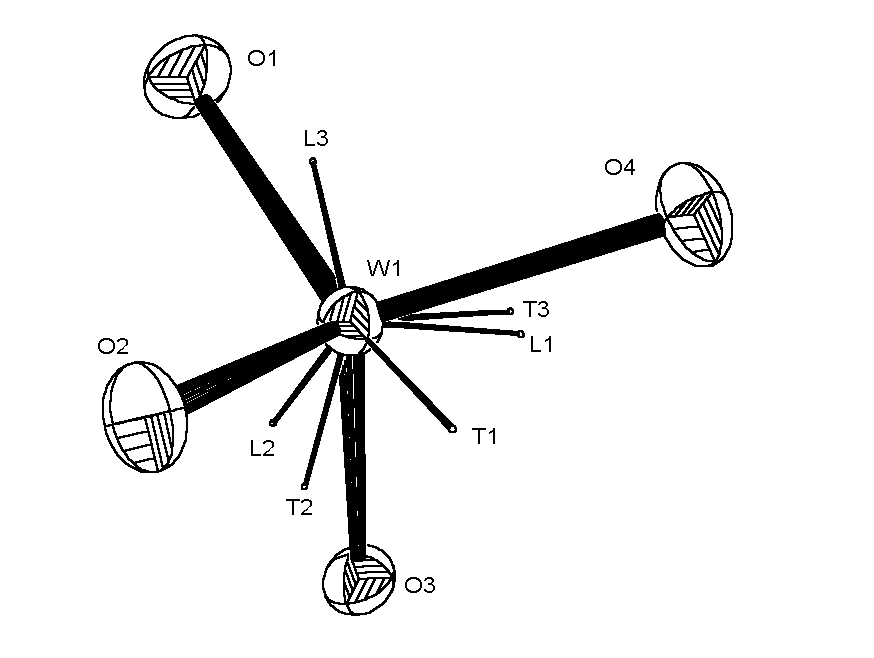
|
ORTEP view of WO4 anion with DIFABS correction,
showing the principal axes of the libration and translation tensors T and L.
|
|
| Table 1 Transmission factors |
| METHOD |
T minimum |
T maximum |
Ratio |
| Multiscan | 0.0169 | 0.1202 | 6.03 |
| DIFABS | 0.0219 | 0.1283 | 5.86 |
| XABS2 | 0.0187 | 0.1283 | 6.86 |
| Analytical | 0.0208 | 0.1475 | 7.09 |
| Gaussian | 0.0206 | 0.1479 | 7.17 |
| Psi-scans | 0.0222 | 0.1283 | 5.78 |
| SHELXA | 0.1072 | 0.5722 | 5.33 |
| Table 2 Refinement results |
| METHOD | R(merge) | R(sigma) | R1 | wR2 |
rho(max) | rho(min) | NIEQ* |
| none | 0.1495 | 0.0946 | 0.0989 | 0.2363 |
24.01 | -7.31 | 818 |
| Multiscan | 0.0654 | 0.0408 | 0.0652 | 0.1639 | 10.49 | -4.87 | 303 |
| DIFABS | 0.0632 | 0.0368 | 0.0348 | 0.0945 | 5.84 | -1.81 | 388 |
| XABS2 | 0.1175 | 0.0772 | 0.0606 | 0.1611 | 10.24 | -5.86 | 769 |
| Analytical | 0.0382 | 0.0242 | 0.0208 | 0.0552 | 2.47 | -1.60 | 210 |
| Gaussian | 0.0383 | 0.0242 | 0.0210 | 0.0555 | 2.41 | -1.59 | 210 |
| Psi-scans‡ | 0.0663 | 0.0387 | 0.0371 | 0.1063 | 4.99 | -3.42 | 342 |
| SHELXA | 0.0817 | 0.0492 | 0.0501 | 0.1316 | 11.73 | -2.74 | 549 |
* NIEQ = number of inconsistent equivalent reflections flagged
by SHELXL-93
‡ No theta correction, unit weights
|
Table 3 Eigenvalues (Å2) of the Uij
tensors for the W and Na atoms*
|
| METHOD | W | Na1 | Na2 |
| | L1 | L2 | L3 | L1 | L2 |
L3 | L1 | L2 | L3 |
| none | 0.0175 | 0.0096 | 0.0082 | 0.0267 | 0.0208 | 0.0148 | 0.0311 | 0.0163 | 0.0134 |
| Multiscan | 0.0188 | 0.0168 | 0.0149 | 0.0300 | 0.0253 | 0.0206 | 0.0277 | 0.0242 | 0.0231 |
| DIFABS | 0.0131 | 0.0124 | 0.0107 | 0.0245 | 0.0202 | 0.0168 | 0.0240 | 0.0193 | 0.0174 |
| XABS2 | 0.0107 | 0.0099 | 0.0062 | 0.0202 | 0.0167 | 0.0151 | 0.0213 | 0.0185 | 0.0116 |
| Analytical | 0.0148 | 0.0134 | 0.0101 | 0.0241 | 0.0200 | 0.0196 | 0.0252 | 0.0215 | 0.0158 |
| Gaussian | 0.0148 | 0.0134 | 0.0101 | 0.0241 | 0.0199 | 0.0197 | 0.0251 | 0.0215 | 0.0158 |
| Psi-scans | 0.0154 | 0.0128 | 0.0069 | 0.0267 | 0.0228 | 0.0113 | 0.0255 | 0.0197 | 0.0145 |
| SHELXA | 0.0167 | 0.0110 | 0.0101 | 0.0249 | 0.0211 | 0.0171 | 0.0270 | 0.0199 | 0.0158 |
* The standard uncertainties on the eigenvalues are ca 0.0002 for
W and ~0.001 for Na
|
Table 4 Bond lengths (Å) for the WO4 anion.
|
| METHOD | W-O1 | W-O2 | W-O3 | W-O4 |
| none | 1.760(8) | 1.776(9) | 1.780(7) | 1.781(5) |
| analytical | 1.760(3) | 1.774(3) | 1.778(3) | 1.787(2) |
| DIFABS | 1.756(4) | 1.782(4) | 1.776(4) | 1.786(2) |
References
- E.N. Maslen in International Tables for Crystallography
Vol C (1995) Section 6.3, pp 520-529.
- K.N. Trueblood, H.B. Burgi, H. Burzlaff, J.D. Dunitz, C.M.
Gramaccioli, H.H. Schulz, U. Shmueli and S.C. Abrahams Acta
Crystallogr Sect. A, 1996, 52, 770-781.
- J. de Meulenaar and H. Tompa, Acta Crystallogr., Sect
A 1965, 19, 1014-1018.
- P. Coppens in Crystallographic Computing ed F.R. Ahmed,
S.R. Hall and C.P. Huber, Copenhagen, Munksgaard, (1970) pp 255-270
- R.H. Blessing, Acta Crystallogr., Sect A 1995, 51,
33-38.
- A.L. Spek, Acta Crystallogr., Sect A 1990, 46, C34.
(b) PLATON, A Multipurpose Crystallographic Tool, Utrecht University,
Utrecht, The Netherlands, A. L. Spek, 1998.
- R.H. Blessing, Cryst. Rev. 1987, 1, 3-58. (b) R. H.
Blessing and D. A. Langs, J.Appl. Crystallogr. 1987, 20,
427-428.
- SADABS: Area-Detector Absorption Correction; Siemens Industrial
Automation, Inc.: Madison, WI, 1996.
- H. D. Flack, Acta Crystallogr., Sect A 1974, 30, 569-573.
(b) Flack, H. D. J. Appl. Crystallogr. 1975, 8, 520-521.
(c) Flack, H. D. Acta Crystallogr., Sect A 1977, 33, 890-898.
- A. C. T. North, D. C. Phillips and F. S. Mathews, Acta.
Crystallogr. Sect A, 1968, 24, 351-359.
- N. Walker and D. Stuart, Acta Crystallogr., Sect
A 1983, 39, 158-166.
- S. Parkin, B. Moezzi and H. Hope J. Appl. Crystallogr.
1995, 28, 53-56
- SHELXL Suite of Programs for Crystal Structure Analysis (Release
97-2). G. M. Sheldrick, Institüt für Anorganische Chemie
der Universität, Tammanstrasse 4, D-3400 Göttingen,
Germany, 1998.
{kind=link}