Anharmonic motion in [PPN]+[FeCo(CO)8]-
see P. Macchi & A. Sironi (2004) Acta Cryst A60 502-509
The crystalline form of the anion [FeCo(CO)8]- as its
[PPN]+ salt is characterized by an
interesting dependence on the temperature: at higher T (up to 343 K)
the molecular geometry of the anion is quite close to the gas phase
prediction, with a semi-bridging carbonyl quite asymmetrically disposed
between the two metals (Co-C = 1.81 Å; Fe-C = 2.28 Å). However, at lower
T (down to 28 K) the carbonyl is more symmetrically disposed with a
significant compression of the Fe-C bond (Co-C = 1.84 Å; Fe-C = 2.15 Å).
The strain tensor (see Figure 1) visualizes quite clearly the softer
direction of the crystal (coinciding in fact with Fe-C bond).
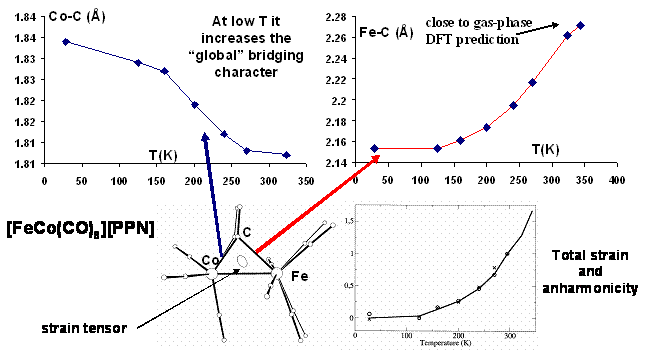 Figure 1
Figure 1
|
The accurate electron density was modeled against X-ray diffraction
data at 125K. This model was used to refine geometrical and thermal
parameters at all temperatures investigated (within the range 28-343 K).
A significant deviation from harmonic motion is expected, especially for
the semi-bridging carbonyl atoms and in fact a Gram-Charlier expansion was
successfully refined (but at the highest T). By means of the program XDPDF,
the probability distribution function can be computed and visualized.
Figure 2 represents the contribution of higher order (3rd, 4th) terms to the
total PDF of the C atom at different temperatures. From 160 K and at least
up to room T, there is a significant contribution of anharmonic motion along
the Fe-C direction that explains the geometric changes occurring and the
softness of that bond. At the highest T, the resolution of the dataset is
significantly reduced (the crystal blows up just 10° above), therefore the
observations collected are of insufficient resolution to allow the fitting
of a proper thermal motion. In fact, the refinement is unstable
and produces a more ambiguous PDF distribution.
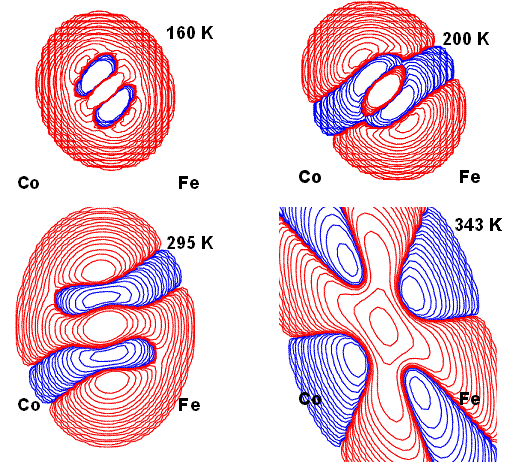 Figure 2
Figure 2
|