Protein
Crystallography: Protein crystallography is especially difficult
because diffraction is often quite weak and because the structures are so complex
with resolution well below the atomic limit. Applications of the ME formalism
in this field have included Avian pancreatic polypeptide, purple membrane using
electron diffraction data, TrpRS and Trytophanyl-tRNA synthetase – all structures
in which traditional direct methods do not work because of the complexity of
the data and its resolution. In these cases the starting phasing were taken
from MIR experiments. The method has been extended to membrane protein data
with the ab initio solution of two membrane proteins in projection at
ca. 9Å resolution, and Beef Liver Catalase at 6 Å.
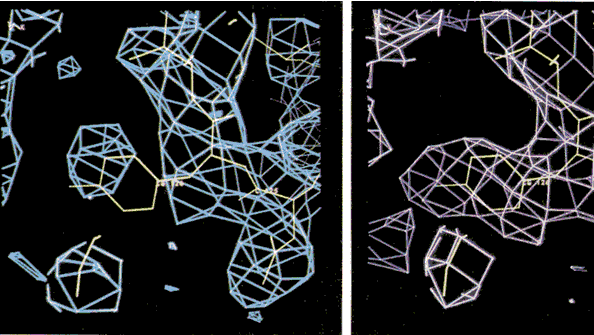
Improving protein electron density maps using maximum entropy.
The groups
interest in the area of protein crystallography uses maximum entropy techniques
in different ways to investigate furthering the use of maximum entropy for
the solution of protein structures.
-
Extrapolating
the resolution range of the observed structure factors. This is the determination
of the reflection intensities which are not measured, but can be determined
using maximum entropy techniques.
-
The
refinement of the protein envelope. The protein envelope is a very low resolution
structure of the protein which defines the boundaries of the protein and
the solvent. It can be obtained from a fragment of the structure and the
observed reflections but it does not always have an accurate shape, therefore
the boundaries of the envelope can be refined using maximum entropy techniques.
-
Solution
of protein structures using protein fragments and envelopes. If a fragment
of a protein can be derived by what ever means, either the heavy atoms or
a partial structure from direct methods, then this information can be used
for the solution of the whole protein. This can be done by incorporting
the most accurate phases of the fragment structure factors into a basis
set, which are then used with maximum entropy to find the solution for the
whole protein.
-
The
use of maximum entropy and envelopes being incorporated into the CCP4 program
ACORN.
Research
is funded by: BBSRC, EPSRC
References
for Protein crystallography
- ‘Direct Methods in Protein
Crystallography – Beef Liver Catalase in its Fully Hydrated Form at Room Temperature’
D.L.Dorset & C.J.Gilmore Acta Cryst. (1999), A55, 448-456.
- 'Direct Methods in Protein
Electron Crystallography: the Ab Initio Structure Determination of Two Membrane
Proteins in Projection using Maximum Entropy and Likelihood’ C.J.Gilmore,
W.V.Nicholson & D.L.Dorset, Acta Cryst. (1996), A52, 937-946.
- 'Trytophanyl-tRNA synthetase
crystal structure reveals an unexpected homology to tyrosyl-tRNA synthetase'
S.Doublié, G. Bricogne, C.J.Gilmore, & C.W.Carter Jnr. Structure
(1995), 3, 17-31.
- 'Overcoming Non-Isomorphism
by Phase Permutation with Likelihood Scoring: Solution of the TrpRS Crystal
Structure' S.Doublié, S.Xiang, C.J.Gilmore, G.Bricogne, & C.W.Carter,
Acta Cryst. (1994), A50, 164-182.
- 'Entropy Maximisation
Constrained by Solvent Flatness: A New Method for Macromolecular Phase Extension
and Map Improvement', S.Xiang, C.W.Carter Jr., G.Bricogne, & C.J.Gilmore,
Acta Cryst. (1993), D49, 193-212.
- 'Phase Extension in
Electron Crystallography Using the Maximum Entropy Method and its Application
to Two-dimensional Purple Membrane Data from Halobacterium Halobium',
C.J.Gilmore, K.Shankland & J.R.Fryer, Ultramicroscopy (1993), 49,
132-146.
- 'A Multisolution Method
of Phase Determination by Combined Maximisation of Entropy and Likelihood.
V The Use of Likelihood as a Discriminator of Phase Sets Produced by the Saytan
Program for a Small Protein' C.J.Gilmore, A.N.Henderson & G.Bricogne,
Acta Cryst. (1991), A47, 842-847.